
모든 iLive 콘텐츠는 의학적으로 검토되거나 가능한 한 사실 정확도를 보장하기 위해 사실 확인됩니다.
우리는 엄격한 소싱 지침을 보유하고 있으며 평판이 좋은 미디어 사이트, 학술 연구 기관 및 가능할 경우 언제든지 의학적으로 검토 된 연구만을 연결할 수 있습니다. 괄호 안의 숫자 ([1], [2] 등)는 클릭 할 수있는 링크입니다.
의 콘텐츠가 정확하지 않거나 구식이거나 의심스러운 경우 Ctrl + Enter를 눌러 선택하십시오.
뇌의 뇌전증
기사의 의료 전문가

최근 리뷰 : 12.07.2025
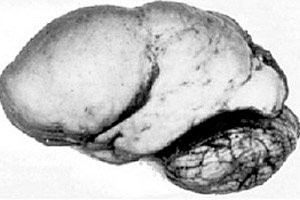
유기성 뇌병리학 중에서 뇌 발달의 선천적 이상이 두드러지는데, 그 본질은 대뇌반구의 피질 표면이 거의 매끈하다는 점, 즉 굴곡과 홈의 수가 부족하다는 점에 있습니다. [ 1 ]
와선이 전혀 없는 경우 무두뇌증(agyria)으로 정의되며, 넓고 편평한 와선이 여러 개 나타나는 경우를 두뇌두뇌증(pachygyria)이라고 합니다. 이러한 결손은 다른 뇌 환원 변형과 마찬가지로 ICD-10 코드 Q04.3을 갖습니다.
역학
희귀질환 통계에 따르면 신생아 10만 명당 1~1.2명의 뇌병변 장애가 발생합니다.[ 2 ], [ 3 ]
일부 자료에 따르면, Miller-Dieker 증후군이 있는 어린이의 경우 고전적 뇌병증의 최대 25-30%가 관찰되며, LIS1 및 DCX 유전자의 점 돌연변이 및 결손은 환자의 거의 85%에서 감지됩니다. [ 4 ]
뇌간증과 관련된 17개 유전자에 대한 유전학 연구에서는 LIS1 돌연변이 또는 결손이 환자의 40%를 차지하고 23%가 DCX 돌연변이와 관련되어 있으며, 그 다음으로 TUBA1A(5%)와 DYNC1H1(3%)이 관련되어 있음을 보여주었습니다.[ 5 ]
원인 리센셥
인간 뇌의 "작업 영역"을 넓히고 중추신경계의 "생산성"을 보장하는 굴곡과 주름 없이 대뇌피질(cortex cerebri)이 형성되는 모든 알려진 원인은 주산기 발달 장애와 관련이 있습니다. 즉, 태아에게 뇌회(lissencephaly)가 발생합니다. [ 6 ]
뇌간증으로 인해 태아의 대뇌 피질 층이 형성되지 않는 것은 뇌간증을 형성하는 신경 세포의 비정상적인 이동이나 이 과정이 조기에 중단된 결과입니다.
대뇌피질 조직 형성에 필수적인 이 과정은 임신 7주차부터 18주차까지 여러 단계로 진행됩니다. 유전적 돌연변이에 대한 민감도가 증가하고 다양한 부정적인 물리적, 화학적, 생물학적 영향이 작용하기 때문에, 정상 범위를 벗어나면 뉴런의 위치가 부정확해지고, 특징적인 구조 없이 피질에 두꺼워진 회백질층이 형성될 수 있습니다. [ 7 ]
어떤 경우에는 어린이의 뇌간증이 밀러-디커 증후군, 워커-바르부르크 증후군 또는 노먼-로버츠 증후군과 관련이 있습니다.
또한 읽어보세요 – 뇌의 발달 결함
병인
모든 뇌회수증(lissencephaly)의 발병 기전이 염색체 이상과 유전자 돌연변이에 기인하는 것은 아닙니다. 그러나 신경모세포와 뉴런이 방사상 아교세포를 따라 정확하게 이동하여 대뇌 피질을 형성하는 데 중요한 역할을 하는 단백질을 암호화하는 유전자들이 알려져 있습니다. 이러한 유전자들의 돌연변이는 이러한 병리학적 기전을 유발합니다. [ 10 ]
특히, 이는 미세소관 다이네인의 세포질 모터 단백질을 조절하는 17번 염색체의 LIS1 유전자와 단백질 더블코르틴(리스뇌팔린-X)을 암호화하는 X 염색체의 DCX 유전자의 산발적 돌연변이(유전 없음)입니다.[ 11 ] 전문가들은 첫 번째 경우를 고전적 리스뇌팔리(1형)로, 두 번째 경우를 X 연관 리스뇌팔리로 정의합니다.[ 12 ]
인산단백질 필라민 1을 인코딩하는 FLN1 유전자가 삭제되면 지시된 신경 이동 과정이 전혀 시작되지 않아 꼬임 현상(무형성)이 전혀 나타나지 않을 수 있습니다. [ 13 ]
CDK5 유전자에서 돌연변이가 발견되었습니다. 이 유전자는 세포 내 대사를 촉진하는 효소인 키나제 효소를 암호화하고, 중추신경계 뉴런의 세포 주기를 조절하며, 태아기 뇌 구조 형성 과정에서 뉴런의 정상적인 이동을 보장합니다.
Norman-Roberts 증후군에서 피질 회전 결함을 유발하는 7번 염색체의 RELN 유전자의 비정상적인 변화는 대뇌 피질 발달 중 신경줄기세포의 이동 및 위치 조절에 필요한 세포외 당단백질 reelin의 결핍을 초래합니다.[ 14 ], [ 15 ], [ 16 ]
ARX 유전자는 전뇌 및 기타 조직에서 중요한 역할을 하는 전사 인자인 비아리스탈렌스 호메오박스 단백질을 인코딩합니다.[ 17 ] ARX 돌연변이가 있는 어린이는 뇌의 일부 결손(대뇌량 무형성), 비정상적인 생식기 및 심각한 간질과 같은 다른 증상을 보입니다.[ 18 ],[ 19 ]
여러 유전자가 뇌병변과 연관되어 있습니다. 이러한 유전자에는 VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A, CDK5가 포함됩니다.[ 20 ]
거대세포바이러스(CMV)는 태아 뇌로의 혈액 공급 감소로 인한 뇌회각증(lissencephaly) 발생과 관련이 있습니다. CMV 감염의 중증도는 재태 기간에 따라 다릅니다. 임신 초기에 신경 세포 이동이 일어나기 때문에 조기 감염은 뇌회각증을 유발할 가능성이 더 높습니다.[ 21 ]
또한, 이 기형의 발생 기전에는 뇌실주위생존영역에서 대뇌피질로의 뉴런 이동이 불완전하거나 나중에 중단되는 것이 포함됩니다. 이러한 경우, 불완전한 뇌회증(lissencephaly)이나 뇌회두증(pachygyria)이 발생하여 여러 개의 넓은 고랑과 회선이 형성되지만(대부분은 존재하지 않음), 이러한 고랑과 회선은 뇌실주위생존영역에서 대뇌피질로의 뉴런 이동이 불완전하거나 나중에 중단되는 것이 포함됩니다.
조짐 리센셥
이 병리의 첫 징후는 (앞서 언급한 증후군이 없는 경우) 출생 직후가 아니라 1개월 반에서 2개월 후에 나타날 수 있습니다. 그리고 가장 흔하게 관찰되는 것은 다음과 같은 뇌회복증의 임상 증상입니다.
- 근육 근력저하증은 종종 경직성 마비와 함께 나타납니다.
- 경련 및 전신성 강직-간대 발작(후두근 긴장의 형태)
- 심각한 정신지체 및 성장 지연
- 신경 및 운동 기능 장애.
삼키는 데 문제가 있으면 아기에게 먹이를 주는 데 어려움이 있습니다. [ 22 ]
심각한 신경운동 장애는 종종 사지마비(사지 전체 마비)로 나타납니다. 손, 손가락, 발가락의 변형도 가능합니다.
1형 뇌수종증이 있는 Norman-Roberts 증후군에서는 두개안면 기형이 관찰됩니다. 심각한 소두증, 낮은 이마 경사 및 튀어나온 넓은 코등, 넓게 벌어진 눈(과두증), 턱의 미발달(소하악증). [ 23 ]
밀러-디커 증후군은 비정상적으로 작은 머리 크기와 넓고 높은 이마, 짧은 코, 관자놀이의 움푹 들어간 부분(양측두 움푹 들어간 부분), 낮게 위치한 기형의 귀를 특징으로 할 수도 있습니다.
심각한 뇌회절증 증후군은 소두증, 즉 안구 크기 감소(소안구증)와 망막 이형성, 폐쇄성 수두증, 그리고 뇌량 결여 또는 저형성 등의 증상이 특징입니다.
합병증 및 결과
이러한 이상으로 인한 합병증으로는 삼키기 기능 장애(연하곤란)와 위식도 역류, 난치성(통제되지 않는) 간질, 잦은 상기도 감염, 폐렴(만성 흡인 포함) 등이 있습니다.
뇌간이 없는 유아는 심방 중격 결손이나 청색증을 동반한 복합 심장 결손(팔로 4징후)의 형태로 선천적 유기성 심장 문제가 있을 수 있습니다. [ 24 ]
대부분의 경우, 출생 후 성장 지연의 결과는 출생 후 24개월 이내에 사망으로 이어진다.
진단 리센셥
진단은 아이의 신체 검사, 부모의 병력, 임신 및 출산 병력에 대한 조사로 시작됩니다.
임신 중에는 무세포 태아 DNA 검사, 양수천자 또는 융모막 융모 샘플링이 필요할 수 있습니다. [ 25 ] 자세한 내용은 선천성 질환의 태아 진단을 참조하십시오.
기기 진단은 뇌 구조를 시각화하고 기능을 평가하는 데 사용됩니다.
- 뇌의 컴퓨터 단층 촬영
- 뇌의 자기공명영상(MRI )
- 뇌파도(EEG). [ 26 ]
임신 중, 20~21주가 지나서 태아를 초음파로 검사하면 뇌의 두정후두엽과 칼카린 홈이 없고, 실비우스열에 이상이 있을 경우, 뇌간 분리증이 의심될 수 있습니다.
감별 진단
선천적 뇌 결손의 다른 증후군과의 감별 진단이 수행됩니다.
20가지 이상의 뇌병변(lissencephaly) 유형이 있으며, 대부분은 고전적 뇌병변(1형)과 자갈뇌병변(cobblestone lissencephaly, 2형)의 두 가지 주요 범주로 나뉩니다. 각 범주는 유사한 임상 양상을 보이지만 유전자 돌연변이는 서로 다릅니다.[ 27 ]
I형 뇌회수증 환자의 뇌 검사 결과, 정상 환자에서 관찰되는 6개 층이 아닌 4개 층으로 구성된 대뇌 피질이 관찰되는 반면, II형 뇌회수증 환자의 경우 대뇌 피질이 무질서하고 덩어리지거나 결절처럼 보이는데, 이는 신경교중배엽 조직에 의해 분리된 피질 뉴런 군집에 의해 대뇌 피질이 완전히 전위되었기 때문입니다. 환자들은 또한 근육과 눈에 이상을 보였습니다.
- 전형적 뇌병변(1형):
- LIS1: 고립성 뇌병변 및 Miller-Dieker 증후군(안면 기형과 관련된 뇌병변). [ 28 ]
- LISX1: DCX 유전자 돌연변이. LIS1 돌연변이로 인한 뇌회상증과 비교했을 때, DCX는 4층이 아닌 6층의 피질을 보입니다.
- 다른 알려진 유전적 결함이 없는 고립성 뇌병증
- 조약돌 뇌병증(2형):
- 워커-바르부르크 증후군
- 후쿠야마 증후군
- 근육, 눈, 뇌의 질병
- 다른 유형은 위의 두 그룹 중 하나에 속할 수 없습니다.
- LIS2: 노먼-로버츠 증후군은 1형 뇌병증이나 밀러-디커 증후군과 유사하지만 17번 염색체가 결손되지 않습니다.
- LIS3
- 리스엑스2
소뇌증: 정상적인 피질 접힘이 없고 머리가 비정상적으로 작은 것이 복합적으로 나타나는 질환입니다. 규칙적인 소뇌증이 있는 아이들은 출생 시 정상적인 크기의 머리를 가지고 있습니다. 출생 시 머리가 작은 아이들은 일반적으로 소뇌증으로 진단됩니다.
또한 뇌 발달 장애의 다른 종류인 뇌병변증과 다발성 뇌회증을 구별하는 것도 중요합니다.
누구에게 연락해야합니까?
치료 리센셥
뇌병변증은 치료할 수 없는 유기적 결함이므로 지지적 치료와 증상적 치료만이 가능합니다. [ 29 ]
우선, 항경련제와 항간질제를 사용하고, 아이가 스스로 삼키지 못하는 경우 위루관을 삽입합니다. 마사지가 효과적입니다.
심각한 수두증의 경우, 뇌척수액을 제거합니다.
예방
전문가들은 미래의 부모가 유전 상담을 받고, 임산부는 적절한 시기에 산부인과에 등록하고 예정된 모든 검사를 받을 것을 권고합니다.
예보
뇌병변 장애 아동의 경우, 예후는 정도에 따라 다르지만, 대부분 아동의 정신 발달은 4~5개월을 넘지 않습니다. 또한, 이 진단을 받은 모든 아동은 심각한 정신운동 장애와 치료가 어려운 간질을 겪습니다. [ 30 ]
NINDS(미국 국립 신경질환 및 뇌졸중 연구소)에 따르면, 뇌병변 장애가 있는 사람들의 최대 수명은 약 10년입니다.